
When it comes to designing medical technology devices, making the Food & Drug Administration (FDA) happy is key. That’s because, if you plan to bring your product to market someday—and I’m sure you do—they’re the final troll under the bridge you’ll need to get past. (In fact, I too need to stay on their good side, so if you could keep that little troll comment between us, that would be great.)
Without any further ado, here are seven MedTech device design pro tips to help you make the FDA happy.
CONDUCTING STUDIES
The FDA likes to see studies—and for good reason! They want to know that your device is both safe and effective, and this can only be demonstrated through rigorous clinical trials. Make sure your team has the expertise necessary to carry out these studies effectively. If they don’t, you’ll want to find an experienced product design partner that does.
Pro Tip #1: Know Your Goal
What are you trying to learn? You can start by gathering as much information about the device as you can. Then, once you know your goal, you can develop a study plan that is both efficient and informative.
Pro Tip #2: Know the Regulatory Deliverables
These can include everything from an IDE application to a 510(k) submission. According to FDA guidance, there are five major pre-validation items and 2 major post-validation items that need to be included in any study.
Pre-Validation Items:
- Updated risk analysis from resulting design changes
- Summary of formative study results/analysis
- Documented design changes and rationale
- A draft of the summative study protocol
- Labeling and IFU to test in the summative study
Post-Validation Items:
- Summative study (validation) protocol and summary
- Summary of total Human Factors effort
Pro Tip #3: Conduct your summative studies BEFORE your large clinicals
This is more of a general guideline than a hard rule, as even the FDA acknowledges that there are exceptions to this order.
Do you need help designing your study? Read our recent guide on the 6 steps for preparing successful MedTech device research.

DESIGN CONTROLS
All medical devices must undergo a rigorous design control process, which helps catch and correct potential problems, ensuring your product is safe and effective. The FDA has outlined specific design control requirements in Section 820.30 of their Quality System Regulation (QSR), and it’s important to adhere to these if you want your product to get approval. Think of this as a sort of guideline for all your development activities leading up to production.
Pro Tip #4: Properly Manage Your Design Controls
The key to any successful design control process is clear documentation, organization, and communication. Make sure all team members are on the same page by creating concise documentation that’s understood by all. It’s also important to use a good software tool to manage your design controls. This will make it easier to track changes, ensure compliance with regulations, and generate accurate reports.
HUMAN FACTORS ENGINEERING
A Human Factors Engineering (HFE) plan will help you think through how people will use your device in the real world and can help you avoid costly redesigns down the road. By integrating HFE activities into development at the start, you can mitigate a ton of issues that could have popped up at the worst possible time.
A portion of this work will be covered in your design controls, which are governed by previously cited guidance documents, as well as the Association for the Advancement of Medical Instrumentation (AAMI) TIR59:2017.
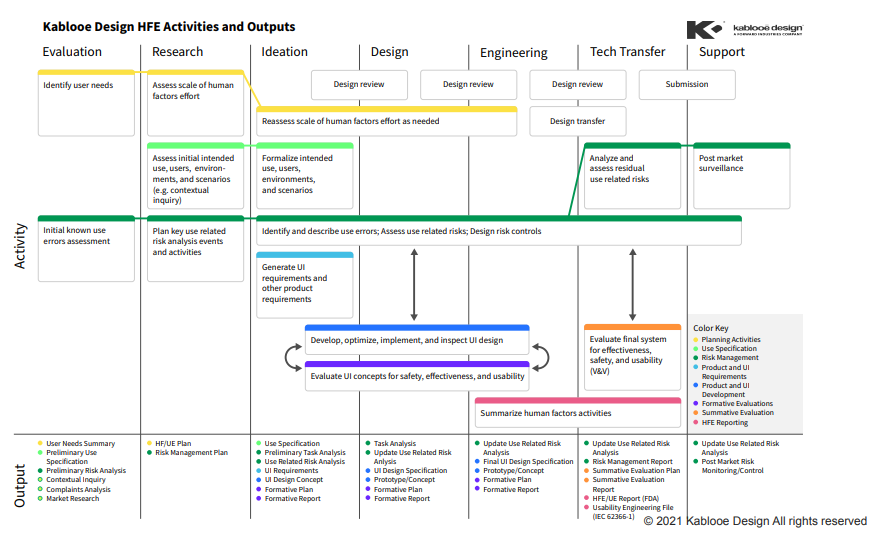
Pro Tip #5: Provide Your HFE Data BEFORE Your summative
The FDA wants to help you evaluate and form your summative protocol to make it more effective and with a higher chance of success—so let them!
Pro Tip #6: Avoid Human Factors Engineering Altogether
I know, I know, this is contradictory to everything I just said. The thing is, though, if your usability risk assessment can show that HFE is not needed, you should make a good case for that and present it to the FDA. This can save you a lot of time and resources in the long run.
US VS. EU MARKETS
The FDA is not the EU, but it is close! The FDA and the European Medicines Agency (EMA) have different approval processes, so it’s important to understand the specific requirements of each one. Differences can include the types of studies required and the level of detail that needs to be included in your submission.
Here are some standards documents for reference:

Pro Tip #7: Understand the Regulatory Differences That Are Most Likely to Affect You
There are three main differences that are important enough to highlight here. Keep in mind, though, that these are general differences and not hard and fast rules. Things change fast.
- The European system asks for a “User Interface Specification”. The US guidance describes tasks to be done but stops short of requiring a specific report for those tasks that align.
- The US systems seem to need to see a lot of summative preliminary work. Europe, on the other hand, is more focused on the summative report.
- The US system puts a lot of focus on analysis such as uFMEA and FTA. The purpose of a uFMEA evaluation is to assess aspects of the user interface as well as task failure effects. An FTA examines assumptions to see whether they are reasonable or if modifications should be made.
ARE YOU READY TO MAKE THE FDA HAPPY?
Sure you are! By following these seven pro tips, you’ll be well on your way to success. After all, making the FDA happy is essential for any MedTech company looking to bring their device to market.
Want one final pro tip? Pre-submission meetings are really important to the FDA, so make sure you work them into your development timeline!
There you have it. If you’re looking for more information on medical device design or if you need help conducting clinical trials or submitting a regulatory application, please contact Kablooe Design today. Our team of seasoned industry experts would be more than happy to assist you.